Control-FREEC: Prediction of copy number alterations and loss of heterozygosity using deep-sequencing data
FAQ page
!!! If you don't see an anwser to your question here, write me an email. Please attach your config file and the complete output of Control-FREEC into the command line.
Q: "Hi, it looks like under Ubuntu 13.04 (and any versions of Unix that are using gcc >= 4.7) FREEC won't compile"
A (by Rory Kirchner): This is due to them removing includes from the gcc standard library for unistd.h: http://gcc.gnu.org/gcc-4.7/porting_to.html
If you edit ThreadPool.h and add
#include <unistd.h>
then it should be good to go.
Q: "Hi there, something went wrong.."
A: Please, read carefully how to create a config file. It can be that you use an incorrect combination of parameters or some mandatary parameters are missing: Control-FREEC Manual.
Check whether you are using the latest version of Control-FREEC. Also, read what Control-FREEC is writing to the command line before throwing an error.
If still you think you did everything well, send me your config file and the complete output of FREEC into the command line.
-
Q: "I am running FREEC on Whole Genome Seq data and keep getting an error that it was unable to extract reads".
A1: If you are using a SAM/BAM file and your have paired-end/mate-pair data, check whether the file is sorted. FREEC generally accepts unsorted files. But in case your file is sorted, you need to use "mateOrientation=0".
A2: Check whether chromosome names in "chrLenFile" correspond to chromosome names in your file with reads. Don't forget that "chrLenFile" should be a tab-delimited file (don't use spaces).
Q: "How to set up the « coefficientOfVariation »?"
A: Parameter « coefficientOfVariation » is used only when you dont provide window size. To use « coefficientOfVariation », comment or delete « window=... » from your config and you will get window size evaluated by Control-FREEC. The default value of « coefficientOfVariation » is 0.05. However, I often use something like « coefficientOfVariation=0.062 » to get smaller windows.
Q: "If I dont have a matched normal could I still use the pileup files to get the genotypes?"
A: Yes, you can. But not for exome-seq and targeted sequencing data.
Q: "Does -1 mean in percentage of uncertainty of the predicted genotype?"
A: -1 means « information is not available ».
Q: "The lower the number in percentage of uncertainty the more reliable the genotype is?"
A: The lower the uncertainty, the more reliable is the prediction. The maximal value should be 100. Values lower than 1 are usually trustable.
Q: "Are the mappability values used when uniqueMatch is FALSE? I'm not sure from the description of the « uniqueMatch » in your tutorial".
A: Even when « uniqueMatch=FALSE », FREEC can use mappability. It simply does not consider regions below the mappability threshold (like pericentromeric or telomeric regions) and makes a guess for them using neighboring mappable regions. The output is much less noisy if you do so.
If « uniqueMatch=TRUE », for the remaining (mappable) regions, FREEC adjusts read count using mappability values. For example, you observe 100 reads in a window with 80% mappable positions. Then, FREEC will consider that in reality in this window there are 100/0.8=125 reads. In theory it seems to be a good approach, but in practice I saw that it may add some unnecessary noise.
Q: "I received the following compilation error: error: access was not declared in this scope".
A: If you are compiling under windows, uncomment "#include < inistd.h >" in the SVfinder.h. If you are under Windows, uncomment "#include "io.h" in the same file.
Q: "The result changes (slightly) depending on whether FREEC reads GC-content from a file or recalculates it anew. Why does it happen?".
A: GC-content is stored as 'float' variables. Issues due to precision of 'float's and rounding result in slight changes in GC-content and thus in slight changes in normalized ratio values (error < 0.001)). Since segmentation algorithm is sensitive to the number of identical values it may (rarely) produce more breakpoints when reading GC-content from a file.
Q: "How is *_mpileup_CNV different from *mpileup_normal_CNV? and depending on the file I use my R plots are so different! why would this be?".
A: *_normal* files correspond to the "normal" genome in case you use one. In the case of exome-seq data, the *_normal* files for copy numbers and ratio values can look strange since they will contain only "2" as copy number predictions. This is because I suppose that copy number of all regions in the normal genome is 2 and I use it to normalize the data from the tumor genome. Thus, there is no point to plot copy numbers for for the normal genome in the case of exome-seq data.
Q: "I'm not sure if GATK ReduceReads could be actually beneficial if the sequencing purpose is to show CNVs, especially if the coverage is so small. Do you think I should avoid it before running Control-FREEC?".
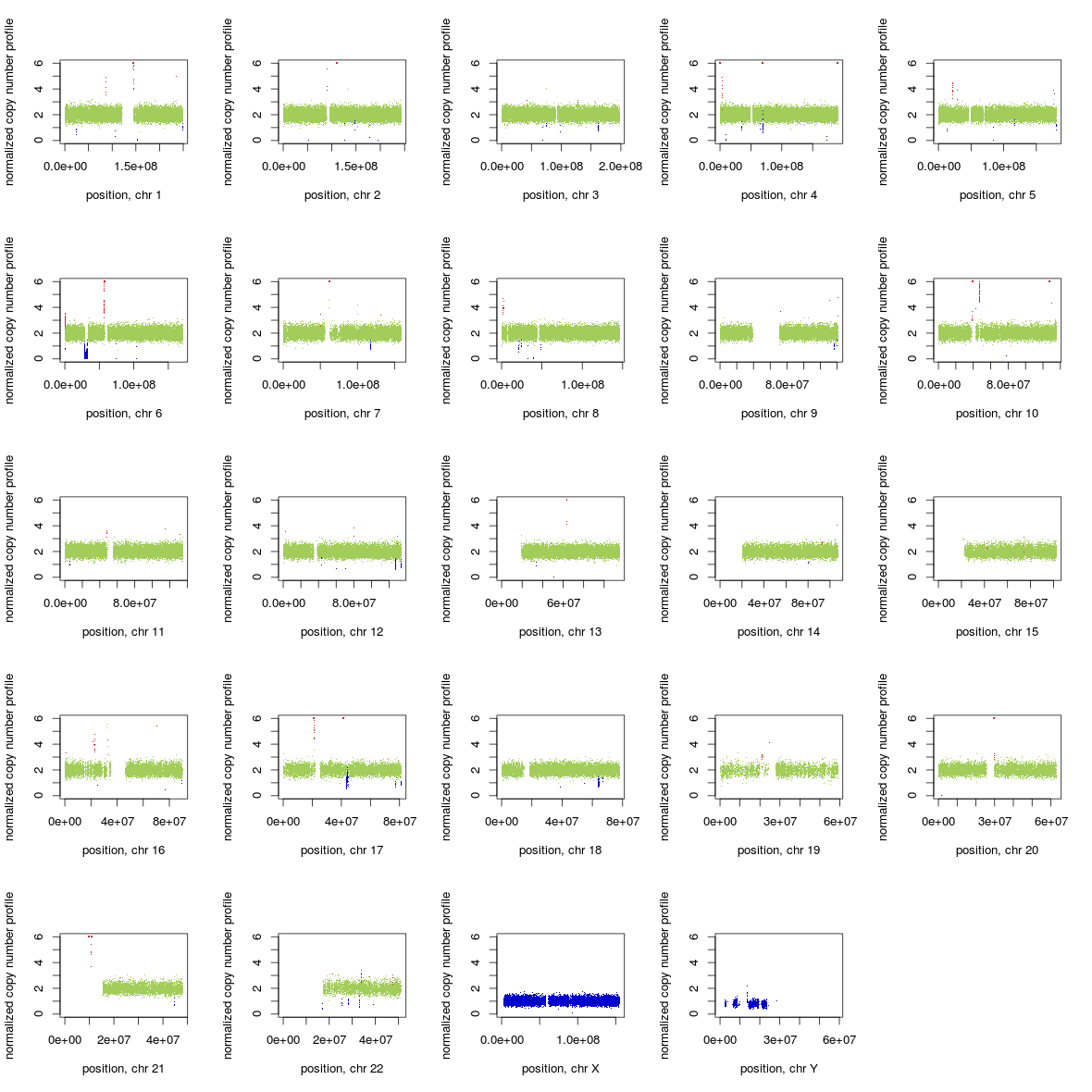
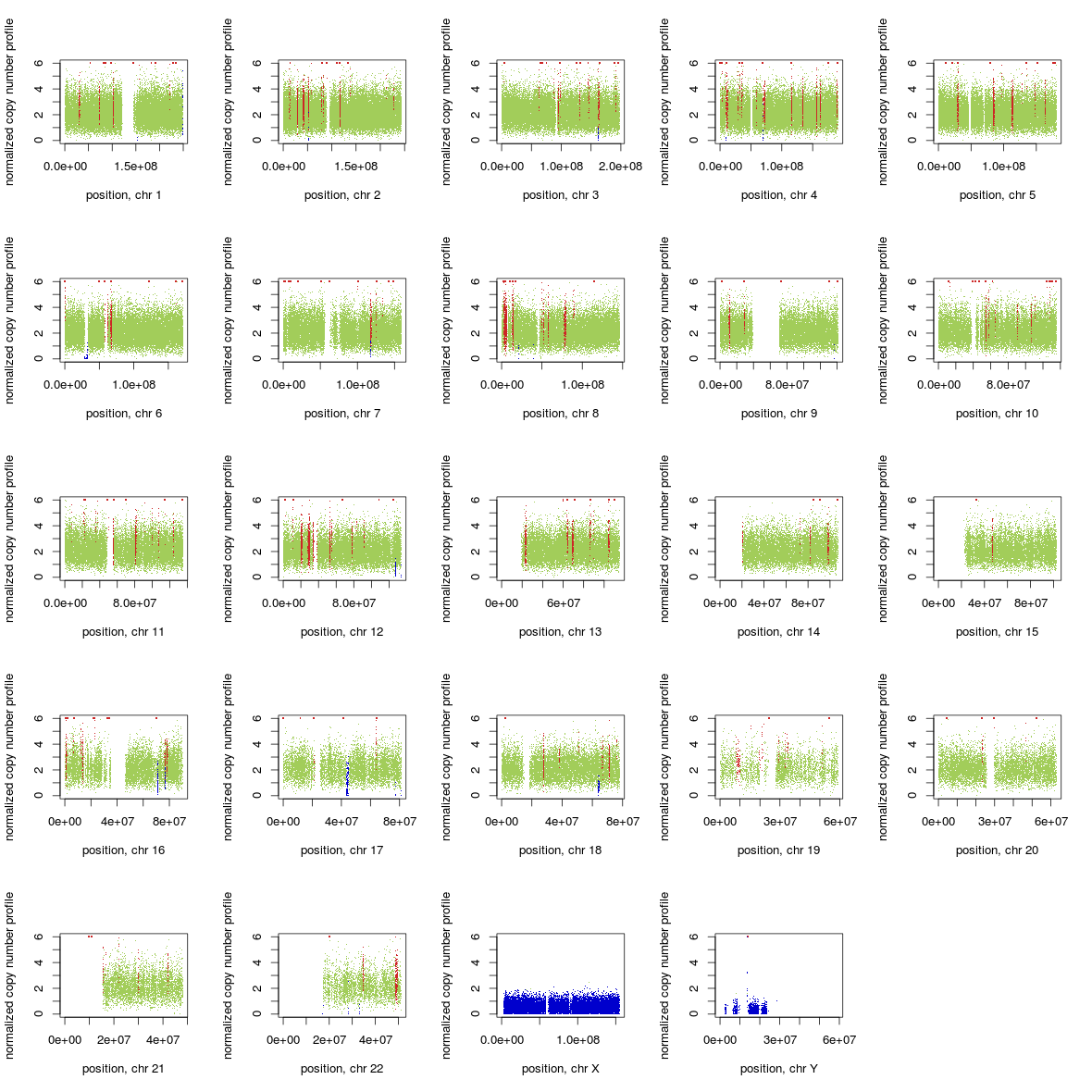
A (by Chiara Rasi): If you use GATK ReduceReads on a BAM file it can affect heavily the performance of Control-FREEC. See the whole genome profile (Illumina whole genome seq 5X) of the same individual, same experiment, with a BAM file before and after using GATK ReduceReads.
Q: "I have used - cat makeGraph.R | R --slave --args 1 some.strange.bug.bam_ratio.txt, the figure looks like a hedgehog".
A : In the case your genome is not a human genome and you don't have chromosome names 1:22 and X and Y, open "makeGraph.R" with a text editor and change chromosome names. Chromosome names in the "makeGraph.R" script should correspond to the chromosome names in the .ratio.txt file. See also "Run FREEC on a something which is not Human"
Q: "Can I supply the data as a CPN file + a pileup of only SNP positions to the program as is?"
A : You can. Simply use both the "mateFile" and "mateCopyNumberFile" parameters. Step and window size must be the same in the new config file and in the config file you used to calculate profile .cnp you are going to provide to "mateCopyNumberFile".
Q: "Where can I find more information about the ways to interpret the results of the graphs (after makeGraph.R)?"
A : Here. In general, blue are losses, red are gains, normal copy number is green. For the BAF profiles: AB and AA/BB (BAF=0.5) is orange, otherwise it's blue. If you uncomment a couple of lines in "makeGraph.R" you could see predicted copy number in black and segment medians in purple.
Q: "Can I run Control-FREEC on RNA-seq data?"
A : No, you should not do this.
Q: "What is the minimal coverage in order to obtain descent CNV results from FREEC (whole genome data)?"
A : Results do not depend on coverage, but the resolution does. You can let FREEC automatically select window size based on coverage by commenting "#window=..."
Then, you can round the evaluated window size and rerun FREEC (say, 431 round to window=500).
Q: "What is the best window size for exome-seq data?"
A : Versions < v8.0: We optimized FREEC for window size "window=500" and "step=250".
Versions > v8.0: Use "window=0" to calculate read count per exon.
Q: "I want to run FREEC with the BAF mode on. I used vcf file generated by GATK. FREEC gives me identical result to the BAF off mode, i.e. there is no BAF calculation at all. I wonder where I got wrong and how to fix it".
A : If the result is identical with and without "[BAF] SNPfile=", then the "SNPfile" format is wrong or you are using an old version of Control-FREEC.
VCF files are accepted only from v9.3.
For previous versions of Control-FREEC (< v9.3), you need to provide a file in a special FREEC format. To create a SNP file in such a format (e.g. hg19_snp131.SingleDiNucl.1based.txt) you need to download a file with SNPs from the UCSC genome browser (Tools => Table Browser), from "Variation and Repeats"/"All SNPs" table. And keep columns 2, 4, 10, 7, 8 and 5. And then keep only entries with "genomic single". Alternatively, you can download a .vcf file from dbSNP (your favorite build) and transform it into the input file for FREEC with vcf2snpFreec.pl. This is no longer needed starting from v9.3, where you can provide a .vcf.gz file with SNP positions.
Q: "Can the FREEC software package be installed on a Mac OS X i7? I've been attempting to build the software from the Linux 64bit source code and have come close. But I receive the error: 'values.h' file not found.".
A : Please open myFunc.cpp in any text editor and replace <values.h> by <limits.h> To do so, you just need to delete a couple of lines around #include <limits.h>.
Q: "Which options in `samtools pileup` I should use to create a pileup file out of a .BAM file on which controlFREEC is able to operate?"
A : samtools mpileup -f ref.fa aln.bam | gzip -c >myPileup.pileup.gz
{kind=link}
{kind=link}
{kind=link}