Control-FREEC: Prediction of copy number alterations and loss of heterozygosity using deep-sequencing data
Manual
Control-FREEC is a tool for detection of copy-number changes and allelic imbalances (including LOH) using deep-sequencing data
originally developed by the
Bioinformatics Laboratory of Institut Curie (Paris). Nowdays, Control-FREEC is supported by the team of
Valentina Boeva at Institut Cochin, Inserm (Paris).
Control-FREEC automatically computes, normalizes, segments copy number and beta allele frequency (BAF) profiles, then calls copy number alterations and LOH.
The control (matched normal) sample is optional for whole genome sequencing data but mandatory for whole exome or targeted sequencing data.
For whole genome sequencing data analysis, the program can also use mappability data (files created by
GEM).
Starting from version v8.0, we provide a possibility to detect subclonal gains and losses and evaluate the likeliest average ploidy of the sample. Also, the procedure for evaluation of tumor purity has been improved.
Starting from Control-FREEC v5.0, the program can be used on exome-sequencing data. Starting from version v8.0, read counts are calculated by exon and not per window (set "window=0").
Starting from Control-FREEC v6.0, the user can use multiple threads to run Control-FREEC. 30x coverage WGS data with a control (i.e., two pileup.gz files) will be fully processed (CNA and LOH info) in one hour using 6 threads.
Input for CNA detection: aligned single-end, paired-end or mate-pair data in SAM, BAM, SAMtools pileup.
Control-FREEC accepts .GZ files. Support of Eland, BED, SOAP, arachne, psl (BLAT) and Bowtie formats has been discontinued starting from version v8.0.
Input for CNA+LOH detection: There are two options: (a) provide aligned reads in SAMtools pileup format. Files can be GZipped; (b) provide BAM files together with options "makePileup" and "fastaFile" (see How to create a config file?)
Output: Regions of gain, loss and LOH, normalized copy number and BAF profiles.
Control-FREEC publications
- Control-FREEC: a tool for assessing copy number and allelic content using next generation sequencing data. V. Boeva, T. Popova, K. Bleakley, P. Chiche, I. Janoueix-Lerosey, O. Delattre and E. Barillot.
Bioinformatics, 2012, 28(3):423-5. PMID: 22155870.
CNA detection part of Control-FREEC (simply FREEC)
- Control-free calling of copy number alterations in deep-sequencing data using GC-content normalization.
V. Boeva, A. Zinovyev, K. Bleakley, J.-P. Vert, I. Janoueix-Lerosey, O. Delattre and E. Barillot. Bioinformatics, 2011, 27(2):268-9.
PMID: 21081509.
LOH detection part of Control-FREEC
Control-FREEC HOWTOs:
- Install the Control-FREEC package
- Run Control-FREEC on a test data
- Create a Config file
- Read Control-FREEC's output
- Calculate significance of Control-FREEC predictions with an R script (new!)
- Visualize Control-FREEC's output
- Translate Control-FREEC's output into Bed or Circos formats
- Generate a GC-content profile without running Control-FREEC
- Run Control-FREEC on something which is not Human:


Contact
Valentina Boeva: valentina.boeva [AT] inserm.fr
Phone: +33.1.44.41.23.89
Distributions:
Control-FREEC is free software; you can redistribute it and/or modify it under the terms of the GNU General Public License as published by the Free Software Foundation (www.fsf.org); either version 2 of the License, or (at your option) any later version.
Test data
- Human data (hg18) for HCC1143 and HCC1143-BL (from Chiang et al., 2009) [ download, 146 Mb ]
- Human data (hg19) for a tumor chromosome 19 (unpublished, to use to test the LOH detection component of Control-FREEC) [ download, 1334 Mb ]
Installation:
- Requirements:
- 10Gb of RAM
- Linux or MACS OS; only older versions of Control-FREEC (up to v6.2) are available for Windows
- R installed
- samtools installed if the input files are in .BAM format
- bedtools installed if you wish to create minipileup files from .BAM
- sambamba installed if you wish to speed up reading of .BAM files
- Download the latest release of Control-FREEC: Linux 64-bit version.
[Starting from Control-FREEC v5.7 Windows is no longer supported. But just in case, we compile Control-FREEC v6.2 for you: Control-FREEC v6.2 for Windows 32-bit . Also, you can still download Control-FREEC v5.6 for Windows 32-bit or contact me for support].
- Unpack Control-FREEC:
type
unzip YOUR_FILE.ziportar -zxvf YOUR_FILE.tar.gz - The Win32 and Linux64 archives include binary versions of Control-FREEC. If you are using Linux,
type
makefrom the Control-FREEC directory to compile the program (change Makefile by removing 64bit-tags if you are running 32bit version of Linux). - Download mappability tracks if you want to include mappability information:
- up to 2 mismatches, hg38, read length 100bp
- up to 2 mismatches, hg19, read length 35-76bp
- up to 2 mismatches, hg19, read length 100bp
- up to 2 mismatches, hg18
- up to 2 mismatches, mm9
- up to 4 mismatches, mm10, read length 100bp (created by Kim Wong)
Do not forget to extract files from the archive! You can also generate a mappability track for other genomes using GEM. - Download files with SNPs (only if you have high coverage data and you want to detect allelic status; then, you must transform read files into pileup format)
- dbSNP151.hg38-commonSNP_minFreq5Perc_with_CHR.vcf.gz
- hg19_dbsnp142_with_CHR_minFreq05.vcf.gz
- hg19_snp131.SingleDiNucl.1based.txt
- hg19_snp137.SingleDiNucl.1based.txt.gz (created by Niklas Malmqvist) Unzip it before the use!!
- hg19_snp142.SingleDiNucl.1based.txt.gz Unzip it before the use!!
- hg18_snp130.SNP.1based.txt
- mm10_snp137 (created by Kim Wong)
Starting from Control-FREEC v9.3, .txt.gz, .vcf and .vcf.gz files are also accepted! For the .txt files with SNPs, please refer to FREEC FAQ Q19 to understand how these files are generated.
Starting from Control-FREEC v10.6, Control-FREEC can work on exome-seq data without a control sample.
Running Control-FREEC on a test data
- To run Control-FREEC download and unzip:
- a test dataset for HCC1143 and HCC1143-BL (from Chiang et al., 2009): test.zip (146 M) or
- a test dataset for tumor chromosome 19 (unpublished,
use it to test LOH predictions by Control-FREEC): testChr19.zip (1334 M)
- Follow the instructions from the README file.
PathToFREEC/freec -conf myConfig.txt -sample sample.bam -control control.bam
or
PathToFREEC/freec -conf myConfig.txt (if BAM files are provided directly in the config file)
Creating a Config file
There should be 2-5 groups of parameters in the config file: [general], [sample], [control], [BAF] and [target]. The last three can be empty if there is no control dataset available, if you do not intend to calculate BAF profiles and call genotypes, or if you are not running the program on targeted data (e.g. Whole Exome Sequencing).- [general]: here you describe all general parameters. Most of them are optional; their default value is indicated below.
- [sample]: here you provide a path to the sample input file and indicate its type (e.g., bam, sam, pileup).
- [control]: here you provide a path to the contol input file and indicate its type. You may have this group empty if you do not have a control dataset.
- [BAF]: here you provide paths to files and parameters to calculate B allele frequency (BAF) profiles and detect LOH regions.
- [target]: here you can provide a .bed file with coordinates of probes, exons or amplicons when you run Control-FREEC on exome sequencing data or other targeted sequencing data. Set "window=0" in the [general] to use read count "per exon" instead of "per window".
[general] group of parameters:
| parameter | description | possible values |
|---|---|---|
| BedGraphOutput | set "BedGraphOutput=TRUE" if you want an additional output in BedGraph format for the UCSC genome browser | Default: FALSE |
| bedtools | path to bedtools (used only to create .pileup files for WES data) | Default: bedtools |
| breakPointThreshold | positive value of threshold for segmentation of normalized profiles | Default: 0.8 use something like 0.6 to get more segments (and thus more predicted CNVs) |
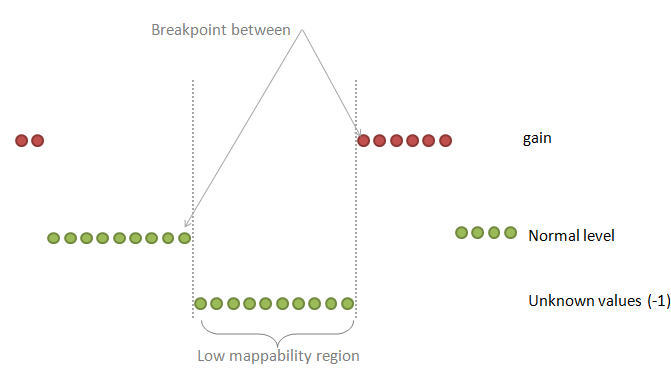
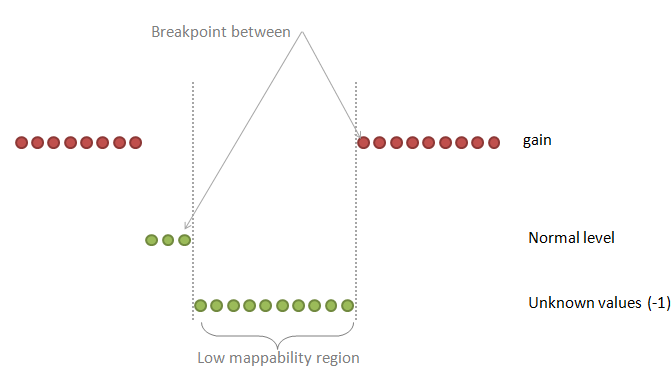
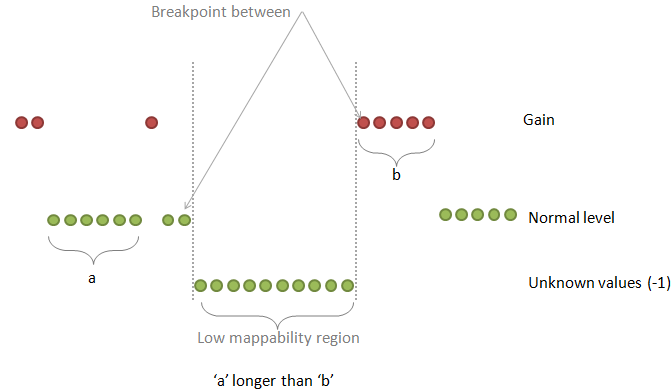
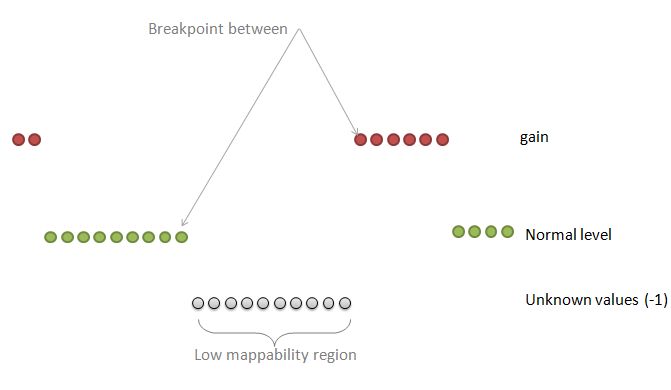
| breakPointType | desired behavior in the ambiguous regions (poly-N or low mappability regions between two different copy number values) |
0: the "unknown" region is attached to the "known" region on the right
1: make a separate fragment of this unknown region and then attaches it to the left or to the right region choosing the longer one 2: make a separate fragment of this unknown region and then attaches it to the left or to the right region but the ploidy copy number has a priority 3: make a separate fragment of this unknown region and then attaches it to the left or to the right region choosing the longer one but this known region should make at least half-size of the unknown region 4: make a separate fragment of this unknown region and do not assign any copy number to this region at all Default: 2 |
| chrFiles | path to the directory with chromosomes fasta files (necessary to calculate a GC-content profile if a control dataset and GC-content profile are not available) | Ex: path/hg19/chromosomes/ |
| chrLenFile | file with chromosome lengths chromosomes that are not in this list won't be considered by Control-FREEC! | Ex: path/hg18.len or
path/hg19.len
(!) files of type hg19.fa.fai are also accepted starting from v9.3 |
| coefficientOfVariation | coefficient of variation to evaluate necessary window size | Default: 0.05 |
| contamination | a priori known value of tumor sample contamination by normal cells | Ex: contamination=0.25 set "contaminationAdjustment=TRUE" to correct for the contamination Default: contamination=0 |
| contaminationAdjustment | set TRUE to correct for contamination by normal cells. If "contamination" is not provided, it will automatically evaluate the level of contamination | Default: FALSE |
| degree | degree of polynomial | Default: 3&4 (GC-content based normalization, WGS) or 1 (control-read-count-based normalization, WES) |
| forceGCcontentNormalization | set to 1 or 2 to correct the Read Count (RC) for GC-content bias and low mappability even when you have a control sample | 0: simply model "sample RC ~ Control RC" 1: normalize the sample and the control RC using GC-content and then calculate the ratio "Sample RC/contol RC" 2: model "sample RC ~ Control RC" bias, and then normalize for GC-content Default (WGS): 0 Default (WES): 1 (≥ v9.5) and 0 (< v9.5) |
| GCcontentProfile | GC-content profile for a given window-size (higher priority than chrFiles) Optional! Necessary only if both a control dataset or chromosome sequences (.fasta for the genome of interest) are not available | Ex: path/GC_profile.cnp |
| gemMappabilityFile | .gem file with information about mappable positions (GEM output) | Ex: out76.gem |
| intercept | intercept of polynomial | Default: 1 - with GC-content, 0 - with a control dataset |
| minCNAlength | minimal number of consecutive windows to call a CNA | Default: 3 for WES and 1 for WGS |
| minMappabilityPerWindow | only windows with fraction of mappable positions higher than or equal to this threshold will be considered (if "gemMappabilityFile" is not provided, one uses the percentage of non-N letters per window) | Default: 0.85 |
| minExpectedGC | minimal expected value of the GC-content for the prior evaluation of "Read Count ~ GC-content" dependency | Default: 0.35 (change only if you run Control-FREEC on a bacterial genome) |
| maxExpectedGC | maximal expected value of the GC-content for the prior evaluation of "Read Count ~ GC-content" dependency | Default: 0.55 (change only if you run Control-FREEC on a bacterial genome) |
| minimalSubclonePresence | detects subclones present in x% of cell population | Default: 100 (meaning "do not look for subclones") Suggested: 20 (or 0.2) for WGS and 30 (or 0.3) for WES |
| maxThreads | number of threads (multi-threading mode) | Default: 1 |
| noisyData | set TRUE for target resequencing data (e.g., exome-seq) to avoid false positive predictions due to non-uniform capture | Default: FALSE |
| outputDir | output directory | Default: "./" (should be an existing folder) |
| ploidy | genome ploidy; In case of doubt, you can set different values and Control-FREEC will select the one that explains most observed CNAs | Ex: ploidy=2 or ploidy=2,3,4 |
| printNA | set FALSE to avoid printing "-1" to the _ratio.txt files Useful for exome-seq or targeted sequencing data | Default: TRUE |
| readCountThreshold | threshold on the minimal number of reads per window in the control sample Useful for exome-seq or targeted sequencing data | Default: 10 recommended value >=50 for for exome data |
| sambamba | path to sambamba (used only to read .BAM files) | Ex: "sambamba = /local/sambamba/sambamba"; Default: will use samtools |
| SambambaThreads | number of threads for Sambamba to use (instead of Samtools) | Defaut: the value of "maxThreads" |
| samtools | path to samtools (used only to read .BAM files or to create .pileup files when option "makepileup" is set) | Ex: "samtools = /local/SAMtools/samtools"; Default: samtools |
| sex | sample sex | "sex=XX" will exclude chr Y from the analysis "sex=XY" will not annotate one copy of chr X and Y as a loss. |
| step | step (used only when "window" is specified); do not use for exome sequencing (instead set "window=0") | Ex: 10000 |
| telocentromeric (!there was a misspeling in all versions up to 6.3) | length of pre-telomeric and pre-centromeric regions: Control-FREEC will not output small CNAs and LOH found within these regions (they are likely to
be false because of mappability/genome assembly issues) 50000 is OK for human/mouse genomes. Use smaller values for yeasts and flies. | Default: 50000 |
| uniqueMatch | Use a mappability profile to correct read counts (in this case a mappability file must be provided with "gemMappabilityFile" ) | Default: FALSE |
| window | explicit window size (higher priority than coefficientOfVariation); | Ex: for whole genome sequencing: "window=50000" for whole exome sequencing: "window=0" |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The parameters chrLenFile and ploidy are required. Either chrFiles or GCcontentProfile
must be specified too if no control dataset is available.
If you provide a path to chromosome files, Control-FREEC will look for the following fasta files in your directory (in this order): 1, 1.fa, 1.fasta, chr1.fa, chr1.fasta; 2, 2.fa, etc. Please ensure that you don't have other files but sequences having the listed names in this directory.
The parameter breakPointThreshold specifies the maximal slope of the slope of residual sum of squares.
This should be a positive value. The closer it is to Zero, the more breakpoints will be called. Its recommended value is between 0.1 and 1.2.
If the parameter gemMappabilityFile is not specified, then the fraction of non-N nucleotides per window is used as Mappability.
To see the effect of using a mappability profile, compare two resuted copy number profiles for NA18507
(a male Yoruba from Ibadan):
without mappability information
and with a mappability profile 35bp up to 2 mismatches, hg19.
At this point, gemMappabilityFile can be used only in the mode without control sample.
[sample] and [control] groups of parameters:
| parameter | description | possible values |
|---|---|---|
| mateFile | file with mapped reads (can be single end reads, mate-pairs or paired-end reads) | Ex: path/sample.bam |
| mateCopyNumberFile | raw copy number profile for a given window-size (higher priority than mateFile) - OPTIONAL (don't need to provide a mateFile if mateCopyNumberFile is provided unless you want to have BAF profiles) | Ex: path/sample.cnp |
| miniPileup | miniPileup file created from the corresponding BAM file dring a previous run of Control-FREEC - OPTIONAL providing this file will significantly speed up the whole process | Ex: path/sample_minipileup.pileup |
| inputFormat | format of reads (in mateFile) | SAM, BAM, pileup, bowtie, eland, arachne, psl (BLAT), BED, Eland. All formated except BAM are also accepted GZipped. |
| mateOrientation | format of reads (in mateFile) | 0 (for single ends), RF (Illumina mate-pairs), FR (Illumina paired-ends), FF (SOLiD mate-pairs) |
Either mateFile or mateCopyNumberFile
must be specified. If you already have read copy number (.cpn) files for mateCopyNumberFile, you can complement it with minipileup file miniPileup or provide mateFile to use it for the [BAF] option. Minipileups are created by Control-FREEC when you run it with the makePileup option of the [BAF] group of parameters.
The parameters inputFormat and mateOrientation are required if mateFile is specified.
If you specify orientation of your reads ("FR","RF", or "FF") then only reads mapping in the corresponding orientation will be used for calculation of copy number profiles.
[BAF] group of parameters:
| parameter | description | possible values |
|---|---|---|
| makePileup | path to a BED or VCF file with SNP positions to create a mini pileup file from the initial BAM file provided in mateFile;if provided, a BAM file can be used for the calculation of BAF instead of .pileup.gz file | Optional; if not used, a pileup file should be provided as mateFile.Such .bed files can be created from hg19_snp142.SingleDiNucl.1based.txt.gz using a command line gunzip -c hg19_snp142.SingleDiNucl.1based.txt.gz | awk {'printf ("%s\t%s\t%s\n", $1,$2-1,$2)'} >hg19_snp142.SingleDiNucl.1based.bed
|
| fastaFile | one fasta file for the whole genome | Optional; need to be provided only when option "makePileup" is used to create a minipileup file from .BAM Ex: path/hg19.fa |
| minimalCoveragePerPosition | minimal read coverage for a position to be considered in BAF analysis | Default: 0 |
| minimalQualityPerPosition | minimal sequencing quality for a position to be considered in BAF analysis | Default: 0; using this option can slow down reading of .pileup |
| shiftInQuality | basis for Phred quality | Default: 0; usually 33 or 64; see fastq quality |
| SNPfile | file with known SNPs | Ex: path/hg19_snp142.SingleDiNucl.1based.txt.gz
.vcf or .vcf.gz files are also accepted in >v9.3 |
[target] group of parameters:
| parameter | description | possible values |
|---|---|---|
| captureRegions | file with capture regions in .BED format | sorted .BED file should contain the following columns:
chr 0-based start 1-based end |
See an example of a config file for whole genome sequencing
here and for
whole exome sequencing here.
Other examples are provided in test.zip and
testChr19.zip.
Control-FREEC's output
_info.txt: parsable file with information about FREEC run (available from v10.3). Information included in the file:
- Program_Version
- Sample_Name
- Control_Used - True if Control-FREEC used a matching control file for data analysis
- CGcontent_Used - True if Control-FREEC used GC-content to normalize read counts
- Mappability_Used - True if Control-FREEC used read mappability data to exclude low mappability regions
- Looking_For_Subclones - True if Control-FREEC was looking for subclonal CNAs
- Breakpoint_Threshold
- Window - 0 if Control-FREEC did not use the window approach but counted reads per exon/amplicon
- Number_Of_Reads|Pairs_In_Sample
- Number_Of_Reads|Pairs_In_Control
- Output_Ploidy
- Sample_Purity - 1 if Control-FREEC did not find contamination or was nor looking for it
- Good_Polynomial_Fit - True if all EM converged
_CNVs: file with coordinates of predicted copy number alterations. Information for each region:
- chromosome
- start position
- end position
- predicted copy number
- type of alteration
- genotype (if [BAF] option is used)
- * percentage of uncertainty of the predicted genotype (max=100). (!) It is **not** the uncertainty of a CNA
- *&** status (somatic or germline)
- *&** percentage of germline CNA/LOH
_ratio.txt: file with ratios and predicted copy number alterations for each window. Information for each window:
- chromosome
- start position
- ratio
- median ratio for the whole fragment resulted from segmentation
- predicted copy number
- * median(abs(BAF-0.5)) per window
- * estimated BAF
- * genotype
- * percentage of uncertainty of the predicted genotype, max=100
- ***** copy number in the subclone
- ***** subclone prevalence
*_BAF.txt: file B-allele frequencies for each possibly heterozygous SNP position. Information for each window:
- chromosome
- start position
- BAF
- fitted frequency of A-allele
- fitted frequency of B-allele
- inferred frequency of A-allele
- inferred frequency of B-allele
- percentage of uncertainty of the predicted genotype, max=100.
_sample.cnp and _control.cnp files: files with raw copy number profiles. Information for each window:
- chromosome
- start position
- number of read starts
GC_profile.cnp: file with GC-content profile. Information for each window:
- chromosome
- start position
- GC-content
- percentage of ACGT-letter per window (1-poly(N)%)
- *** percentage of uniquely mappable positions per window
****_ratio.BedGraph: file with ratios in BedGraph format for visualization in the UCSC genome browser. The file contains tracks for normal copy number, gains and losses, and copy neutral LOH (*). Information for each window:
- chromosome
- start position
- end position
- ratio*ploidy
** if [control] option is used
*** if gemMappabilityFile is used
**** if [general] BedGraphOutput=TRUE (see config file options)
***** if minimalSubclonePresence is used
Calculate significance of Control-FREEC predictions with an R script
One can add the p-values to predicted CNVs by running assess_significance.R:
cat assess_significance.R | R --slave --args < *_CNVs > < *_ratio.txt >
R and package rtracklayer should be installed!
Running this script will add both Wilcoxon test and Kolmogorov-Smirnov test p-values to each line of _CNVs file.
It will also add headers to the columns of _CNVs.
Visualize Control-FREEC's output
awk '$3!=-1 {print}' $outdir/$sample.pileup.gz_normal_ratio.txt > $outdir/$sample.pileup.gz_normal_ratio_noNA.txt
awk '$3!=-1 {print}' $outdir/$sample.pileup.gz_ratio.txt > $outdir/$sample.pileup.gz_ratio_noNA.txt
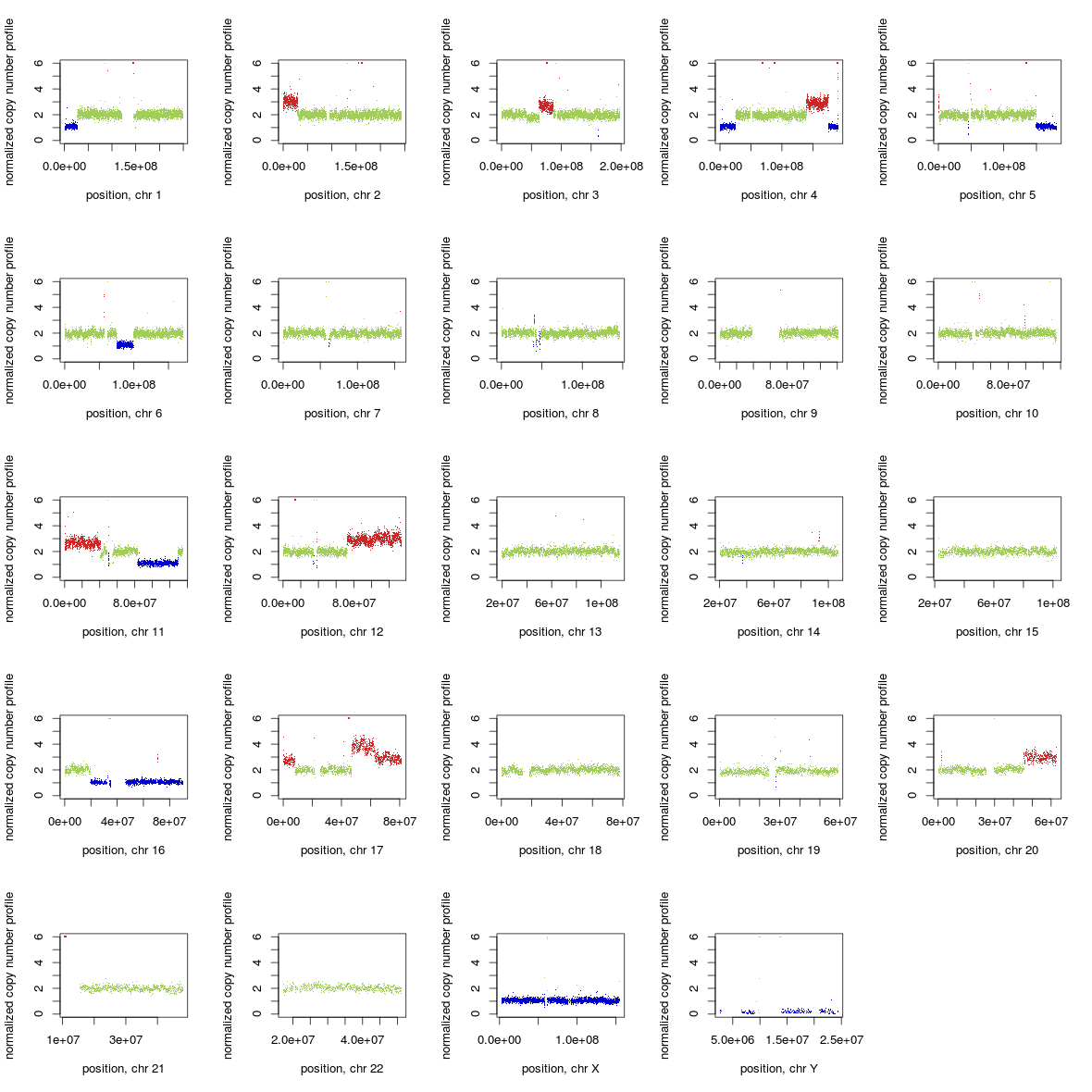
then use *_ratio_noNA.txt files instead of *_ratio.txt in the example below.You can visalize normalized copy number profile with predicted CNAs as well as BAF profile by running makeGraph2.0.R:
cat makeGraph2.0.R | R --slave --args <*_ratio.txt> [<*_BAF.txt>]
R should be installed!
Example of visualization:
Translate Control-FREEC's output into Bed or Circos formats
To translate an output of Control-FREEC to a .BED file (so to view it in the UCSC Genome Browser) or to the Circos format, use these scripts: freec2bed.pl and freec2circos.pl
Generate a GC-content profile without running Control-FREEC
If for some reason you want to separately generate a GC-content profile (GC_profile.cpn) and
run Control-FREEC on your read file, you can use gccount (for Linux, 64bit)
to generate "GC_profile.cpn". You can use the same config file as for the main application. No need to delete or comment anything. However,
window size (option "window") is mandatory. "step" and "gemMappabilityFile" are optional.
Run FREEC on a something which is not Human
The crucial thing to successfully run FREEC on your data is to
- Create a file with chromosome lengths (it can be, for example, mm9.fa.fai)
- Change the "minExpectedGC" and "maxExpectedGC" parameters to fit your data
- Change the value of the "telocentromeric" parameter
- Change chromosome names in "makeGraph2.0.R"
Create a file with chromosome lengths:
If you do not know chromosome lengths for your genome of interest, you can calculate them using this script: get_fasta_lengths.pl
To run this script:
- if you have all your chromosomes in one multifasta file "MyGenome.fa", then type
perl get_fasta_lengths.pl MyGenome.fa - if you have all the chromosomes in separate fasta files in the directory "MyGenome", then type
cat MyGenome/*.fa* > MyGenome.fa
perl get_fasta_lengths.pl MyGenome.fa
rm MyGenome.fa
Change the "minExpectedGC" and "maxExpectedGC" paramters:
For the Drosophila genome set:
minExpectedGC = 0.3 and maxExpectedGC = 0.45.
Also it seems to be necessary to remove all the "Het" chromosomes from the analysis by removing them from the chromosome lengths file (thanks to Joel McManus).
Change the value of the "telocentromeric" parameter:
telocentromeric=50000 is OK for human/mouse genomes. Use smaller values for yeasts and flies.
Do not set anything for exome-seq data.
Change chromosome names in "makeGraph2.0.R":
In the case your genome is not a human genome and you do not have chromosome names 1:22 and X and Y, open "makeGraph2.0.R" with a text editor and change chromosome names. Chromosome names in the "makeGraph2.0.R" script should correspond to the chromosome names in the .ratio.txt file.